Code and software
Analytical tools and pipelines for evolutionary biology research
We use our understandings of natural history to inform the development of more biologically complex and realistic statistical models.
Check out our GitHub Organization: github.org/tribblelab
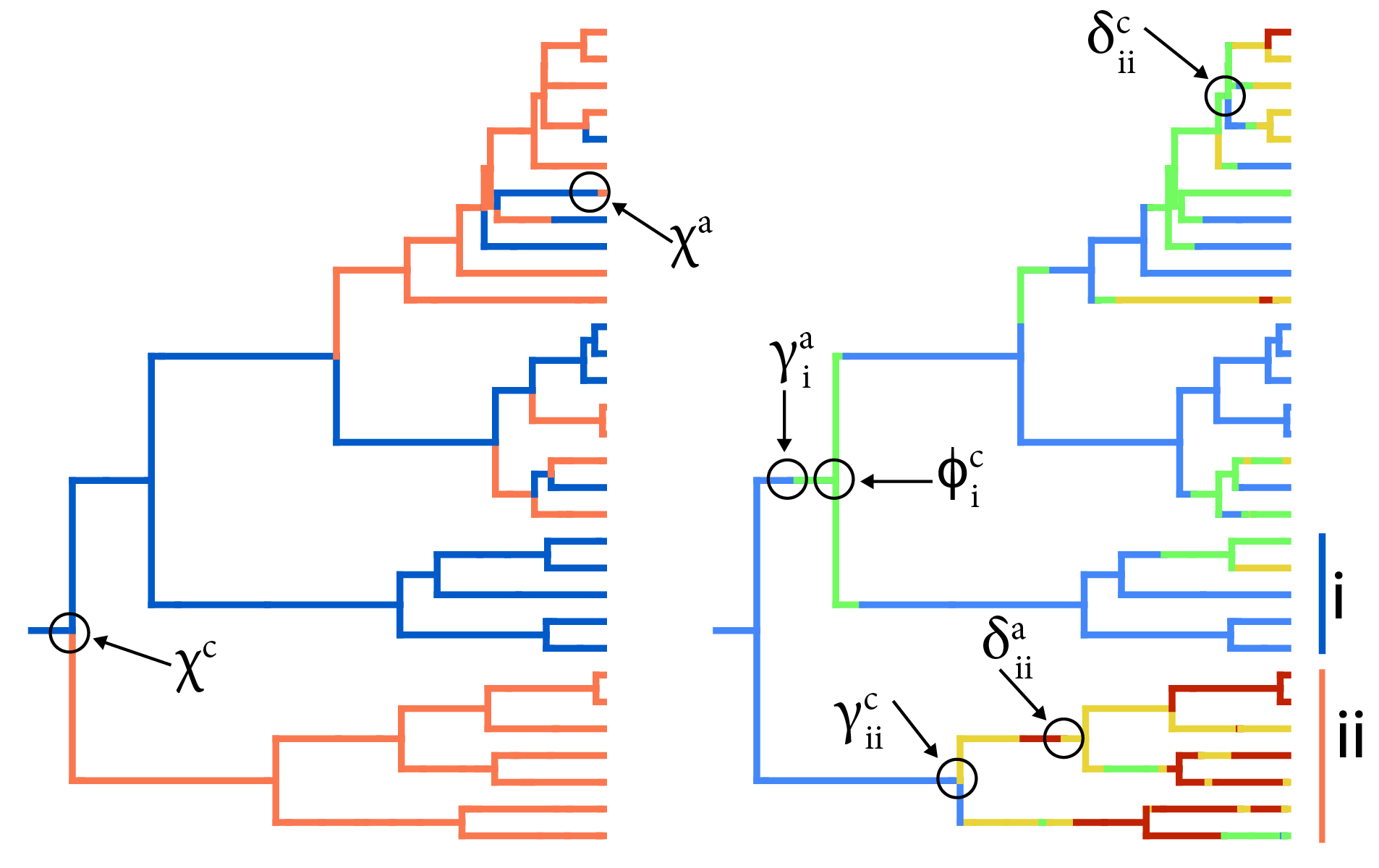
ChromoHiSSE
Our recent work in this area includes the development of cladogenetic chromosome evolution models that account for hidden rate variation (ChromoHiSSE), implemented in RevBayes + TensorPhylo. ChromoHiSSE extends the ChromoSSE model to model different modes of chromosome evolution and diversification across lineages. You can find the code for this project on GitHub.
Discrete-Continuous trait evolution
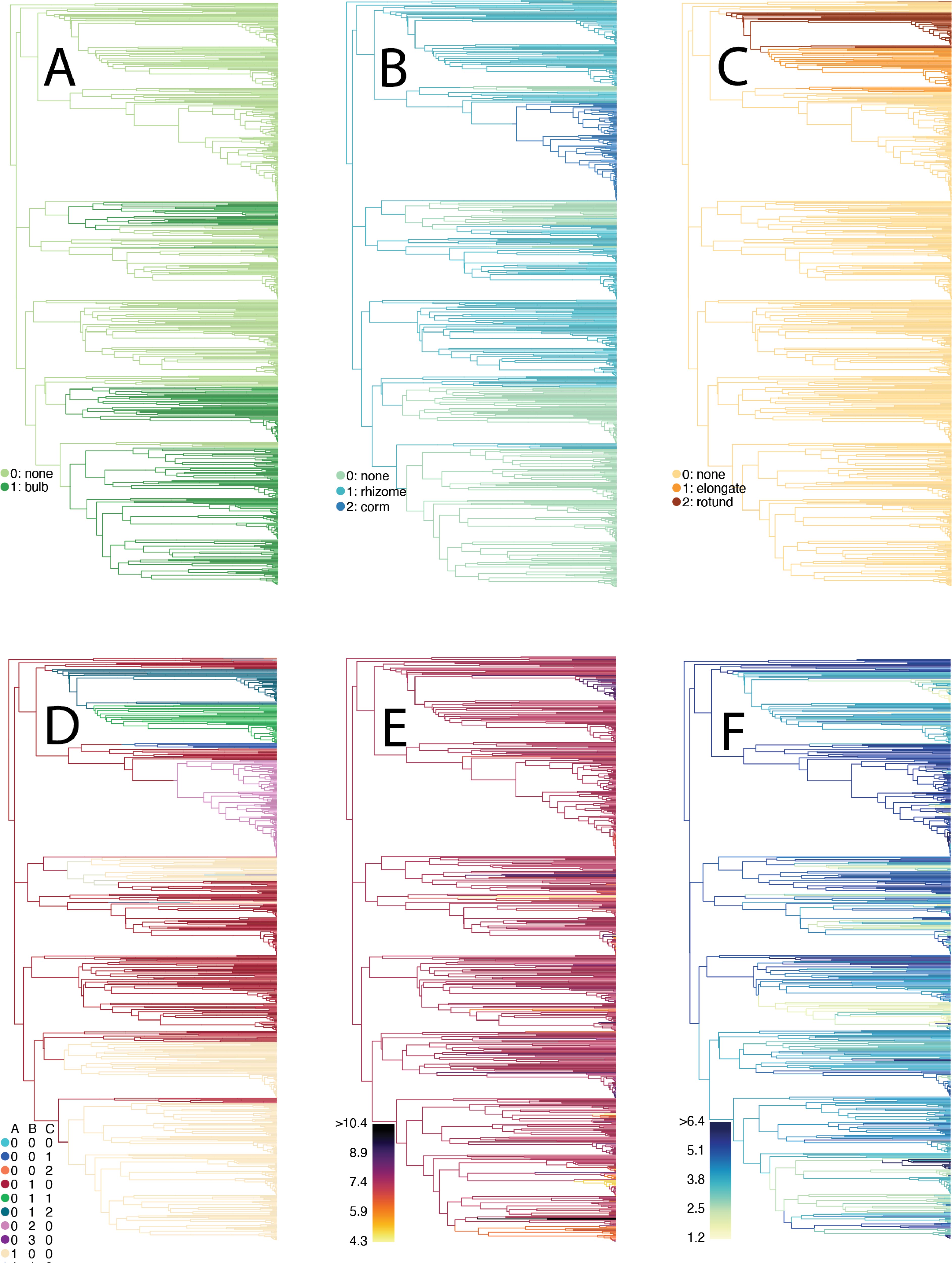 We also developed an analytical pipeline to test for correlated evolution between discrete and continuous traits, while accomodating complex models of discrete trait evolution such as PARAMO.
Our method estimates state-averaged continiuous-trait optimal phenotypes to determine if the optimal contiuous-trait value changes depending on the corresponding discrete state.
We used this pipeline to determine that the tissue type from which underground storage organs are derived significantly affects the plants’ climatic seasonality. You can find the code for this project on GitHub.
We also developed an analytical pipeline to test for correlated evolution between discrete and continuous traits, while accomodating complex models of discrete trait evolution such as PARAMO.
Our method estimates state-averaged continiuous-trait optimal phenotypes to determine if the optimal contiuous-trait value changes depending on the corresponding discrete state.
We used this pipeline to determine that the tissue type from which underground storage organs are derived significantly affects the plants’ climatic seasonality. You can find the code for this project on GitHub.
Comparing diversification rate models
Futhermore, in a collaborative project, we compared empirical diversification rate estimates from multiple popular methods, including BAMM, the Lineage-Specific Birth-Death-Shift process implemented in Revbayes (and its derivitive PESTO), ClaDS2, and the approximate Multi-Type Birth-Death model implemented in BEAST. demonstrating that method choice matters across most phylogenies. We provide a set of scripts for comparing and visualizing the results of analyses across methods on GitHub.
RevGadgets
 We co-develop the R Package RevGadgets, which provides user-friendly processing and plotting functionality for the output RevBayes analyses. We provide a tutorial for RevGadgets on the RevBayes website. The package is available on CRAN and GitHub.
We co-develop the R Package RevGadgets, which provides user-friendly processing and plotting functionality for the output RevBayes analyses. We provide a tutorial for RevGadgets on the RevBayes website. The package is available on CRAN and GitHub.